Die Sahara war auf Besuch, selbst in Wien liegt noch überall der Staub – dabei waren wir gar nicht so sehr betroffen wie einige unserer Nachbarn.


(Tagesaktuelle Karten dazu gibt es von Copernicus)
Aber wie viel Staub ist das eigentlich, der da so herüberströmt aus der Sahara?
Nicht wenig – zwischen 22 und über 23 Tonnen pro Sekunde (84.000 Tonnen pro Stunde) traten am Höhepunkt — am 15.4. mittags — über den 38 Breitengrad zwischen 8 und 28° östlicher Länge.
Die beiden Zahlen stellen einmal den gesamten Staubfluss dar, das andere Mal den “Nettoimport”, jeweils für den gesamten betrachteten Abschnitt – der Nettoimport ist niedriger, da es zu jedem Zeitpunkt zumindest punktuell auch Nordwind gab. Er ist aber nur geringfügig niedriger, da die dann meist weniger Material mitführen als die zeitgleichen Südwinde frisch aus der Sahara in anderen Abschnitten(1).


Das European Centre for Medium Range Weather ForecastsEuropean Centre for Medium Range Weather Forecasts stellt nach einer kostenlosen und unkomplizierten Selbstanmeldung ganz brauchbare Rohdaten zur Verfügung(2). Ich hab mir also für 13.-17.4., jeweils mitternachts und mittags, folgende Datentypen runtergesaugt:
- Staubkonzentration (in drei Subkategorien je nach Korngröße, in Gewichtsanteilen)
- Die Nord-Süd-Komponente der Windgeschwindigkeit (“V component of wind”, als solche in den Rohdaten, erspart mir die Trigonometrie)
- Temperaturen
- “Geopotential Heights”
Die Temperaturen sind notwendig, da sie die Dichte der Luft mitbestimmen, und damit miteinbezogen werden müssen, um von den Gewichtsanteilen Staub (kg/kg) zu Staubkonzentrationen pro Volumen (kg/m3) zu kommen (kg Staub / kg Luft * kg Luft / Kubikmeter = kg Staub / Kubikmeter).
Die “Geopotential Heights” sind notwendig, weil die Schichten in den Rohdaten nicht nach Höhe über dem Boden sondern nach Isobaren aufbereitet sind: Nach Schichten mit gleichem Luftdruck. Um also die Staubkonzentrationen je Höhe zu bekommen, und damit auch die Basis für die Interpolation für alle anderen Höhen, muss ich erst die Geopotential Heights abfragen.
Ich rechne also für jede Lokalität die summierten Staub-Gewichtsanteile je Isobare * Luftdichte je Isobare, um auf die Staubkonzentration je Isobare zu kommen, mulitpliziere diesen Wert mit der Nord-Süd-Komponente de Windgeschwindigkeit je Isobare, um auf die Transportrate über den Breitengrad hinweg zu kommen (in kg pro Meter der gedachten Linie und pro Höhenmeter der Luftsäule). Diese Werte plotte ich gegen die Höhen der jeweiligen Isobare und bestimme auf dieser Basis eine Interpolationsfunktion, die mir für jede Höhe eine geschätzte Staub-Transportrate gibt.
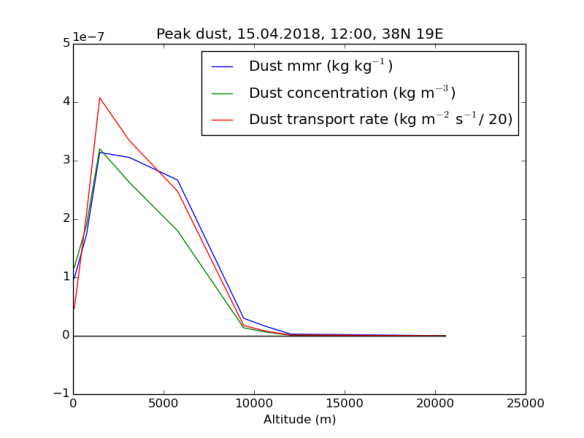
Hier sind diese drei Zahlen — Staub in Masseanteilen, Staub in Masse je Volumenseinheit, und Staubtransport je Höhenmeter je Meter Grundlinie je Sekunde — für den Ort und Zeit der höchsten Transportrate nach Höhenschichten aufgelöst dargestellt. Die blaue Linie stellt den Masseanteil (mass mixing ratio) von Staub an der Luft dar. Man sieht, dass der sich bis auf eine stattliche Höhe von mindestens 6000 Metern im – sehr hohen – Bereich von mehreren hundert Mikrogramm/kg (oder ppbm) bewegt. Die grüne Linie, der Staub je Kubikmeter, sinkt dagegen schon vorher stark ab, da auf diesen Höhen die Luft schon deutlich dünner ist. Die rote Linie stellt die eigentliche Grundlage der Berechnung dar: Den pro Zeiteinheit nach Norden transportierten Staub. Um sie in der gleichen Grafik darstellen zu können, musste ich sie um einen Faktor von 20 herunterskalieren: An diesem Ort herrschten am 15.5. mittags über einen breiten Höhenbereich hinweg Winde mit einer Nordkomponente von 20-25 m/s und mehr vor.
Je Lokalität und Zeitpunkt ergibt sich daraus eine Kurve, die den Materialtransport in nördlicher (positive Werte) oder südlicher Richtung abhängig von der Höhe beschreibt. Hier beispielhaft die entsprechenden Kurven für 15° Ost (östliches Sizilien) und 24° Ost (Athen):


Bereits zu Anfang des untersuchten Zeitraums herrschen in Sizilien Südwinde (mit dem Potential, Staub nach Norden zu transportieren) vor, die allerdings noch verhältnismäßig wenig Staub transportieren. Erst um den 14. April mittags geht die Menge des transportierten Staubs sprunghaft nach oben, am 15. April mittags wird der Höhepunkt erreicht, kurz darauf dreht aber auch schon wieder der Wind (und damit fallen auch die Staubmengen wieder). Anders in Athen, hier herrschen bis zum Morgen des 15. noch Nordwinde vor, erst am 15. dreht der Wind auf Süd, die höchsten Staubwerte werden hier erst am 16. erreicht.
===================
(1) Hier nochmal der jeweilige lokale Netto-Staubtransport aufgedröselt für jeden einzelnen Längendgrad. Man sieht sehr gut, dass es (fast) überall zeitweise Nordwinde gab, die aber nirgends soviel Staub transportierten wie die Südwinde zu anderen Zeitpunkten, oder die zeitgleichen Südwinde an anderen Orten. Die X-Achse sind Messzeitpunkte, die Y-Achse der Staubübertritt pro Stunde pro Kilometer des 38 Breitengrads auf der jeweiligen Länge, negative Werte ein stellen Nordwinde und einen Staubtransport in Richtung Süden dar. Der absolute Höchstwert mit fast 160 Tonnen pro Stunde pro Kilometer wird am 15. 4. mittags am 19 Längengrad erreicht – über dem Ionischen Meer, etwa am halben Weg zwischen Kalabrien und Zakynthos.





















(2) Konkret habe ich verwendet: TIGGE-Daten für GH, Temperatur und Windgeschwindigkeiten, und CAMS für die Staubwerte. Die Installation der notwendigen Software, um mit den Daten dann auch was anzufangen, war nicht ganz so unkompliziert wie der Zugriff auf diese selbst.
(*) Die Berechnungen sind auf Github dokumentiert.